1. Introduction
roteins are molecules with macro structure that are responsible for a wide range of vital biochemical functions, which includes acting as oxygen, cell signaling, antibody production, nutrient transport and building up muscle fibers. Specifically, the proteins are chains of amino acids, of which there are 20 different types, coupled by peptide bonds [2]. The three-tiered structural hierarchy possessed by proteins is typically referred to as primary and tertiary structure. This is because the higher-level and secondary level [1], [2] structures determine the function of the proteins and consequently, the insight into its function can be inferred from that.
As genome sequencing projects are increasing tremendously. The SWISS-PORT databases [3], [4] of primary protein structures are expanding tremendously. Protein Data Banks are not growing at a faster rate due to innate difficulties in finding the levels of the structures. Structure determination [5], [6] procedure experimental setups will be very expensive, time consuming, require more labor and may not applicable to all the proteins. Keeping in view of shortcomings of laboratory procedures in predicting the structure of protein major research have been dedicated to protein prediction of high level structures using computational techniques. Anfinsen did a pioneering work predicting the protein structure from amino acid sequences [6], [7]. This is usually called as protein folding problem which is the greatest challenge in bioinformatics. This is the ability to predict the higher level structures from the amino acid sequence.
By predicting the structure of protein the topology of the chain can be described. The tree dimensional arrangement of amino acid sequences can be described by tertiary structure. They can be predicted independent of each other. Functionality of the protein can be affected by the tertiary structure, topology and the tertiary structure. Structure aids in the identification of membrane proteins, location of binding sites and identification of homologous proteins [9], [10], [11] to list a few of the benefits, and thus highlighting the importance, of knowing this level of structure This is the reason why considerable efforts have been devoted in predicting the structure only. Knowing the structure of a protein is extremely important and can also greatly enhance the accuracy of tertiary structure prediction. Furthermore, proteins can be classified according to their structural elements, specifically their alpha helix and beta sheet content.
2. Related Works in Structure Prediction
The Objective of structure prediction is to identify whether the amino acid residue of protein is in helix, strand or any other shape. In 1960 as a initiative step of structure prediction the probability of respective structure element is calculated for each amino acid by taking single amino acid properties consideration [1], [3], [6]. This method of structure prediction is said to be first generation technique. Later this work extended by considering the local environment of amino acid said to be second generation technique. In case of particular amino acid structure prediction adjacent residues information also needed, it considers the local environment of amino acid it gives 65% structure information. So that extension work gives 60% accuracy. The third generation technique includes machine learning, knowledge about proteins, several algorithms which gives 70% accuracy. Neural networks [10], [11] are also useful in implementing structure prediction programs like PHD, SAM-T99.
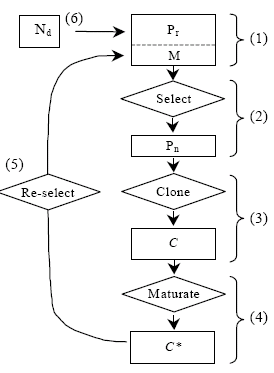
The evolution process is directed by the popular Genetic Algorithm (GA) with the underlying philosophy of survival of the fittest gene. This GA framework can be adopted to arrive at the desired CA rule structure appropriate to model a physical system. The goals of GA formulation are to enhance the understanding of the ways CA performs computations and to learn how CA may be evolved to perform a specific computational task and to understand how evolution creates complex global behavior in a locally interconnected system of simple cells. Artificial immune systems are motivated by the theory of immunology. The biological immune system functions to protect the body against pathogens or antigens that could potentially cause harm. It works by producing antibodies that identify, bind to, and finally eliminate the pathogens. Even though the number of antigens is far larger than the number of antibodies, the biological immune system has evolved to allow it to deal with the antigens. The immune system will learn the criteria of the antigens so that in future it can react both to those antigens it has encountered before as well as to entirely new ones. In 2002, de Castro and Timmis [17], suggested that "for a system to be characterized as an artificial immune system, it has to embody at least a basic model of an immune component (e.g. cell, molecule, organ), it has to have been designed using the ideas from theoretical and/or experimental immunology.
IV. Step 1: Generate a AIS-PSMACA with k number of attractor basins.
3. Design of MACA based Pattern Classifier with Artificial Immune System
Step 2: Distribute S into k attractor basins (nodes).
Step 3: Evaluate the distribution of examples in each attractor basin
Step 4: If all the examples (S") of an attractor basin (node) belong to only one class, then label the attractor basin (leaf node) for that class.
Step 5: If examples (S") of an attractor basin belong to K" number of classes, then Partition (S", K").
Step 6: Stop.
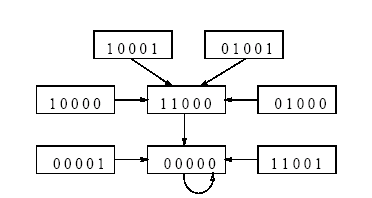
A special class of non-linear CA, termed as Multiple Attractor CA (SPECIAL MACA), has been proposed to develop the model. Theoretical analysis, reported in this chapter, provides an estimate of the noise accommodating capability of the proposed SPECIAL MACA based associative memory model. Characterization of the basins of attraction of the proposed model establishes the sparse network of nonlinear CA (SPECIAL MACA) as a powerful pattern recognizer for memorizing unbiased patterns. It provides an efficient and cost-effective alternative to the dense network of neural net for pattern recognition. Detailed analysis of the SPECIAL MACA rule space establishes the fact that the rule subspace of the pattern recognizing/classifying CA lies at the edge of chaos. Such a CA, as projected in [20], is capable of executing complex computation. The analysis and experimental results reported in the current and next chapters confirm this viewpoint. A SPECIAL MACA employing the CA rules at the edge of chaos is capable of performing complex computation associated with pattern recognition.
4. c) Algorithm Single Point Crossover
Input : Two randomly selected rule vectors (Parent 1 and 2). Output : Resultant rule vectors (Offspring 1 and 2).
Step 1: Randomly generate a number "q" in between 1 and n.
Step 2: Take the first q rules (symbols) from first rule vector (Parent 1) and the (n-q) rules of Parent 2. Form a new rule vector (Offspring 1) concatenating these rules.
Step 3: Form Offspring 2 by concatenating the first q rules of Parent 2 and the last (n-q) rules of Parent 1.
Step 4: Stop.
5. d) Random Generation of Initial Population
To form the initial population, it must be ensured that each solution randomly generated is a combination of an n-bit DS with 2m number of attractor basins (Classifier #1) and an m-bit DV (Classifier #2). The chromosomes are randomly synthesized according to the following steps. V.
6. Experimental Step
? Select the target CA protein (amino acid sequence) T, whose structure is to be predicted.
? Perform a AIS-PSMACA search, using the primary amino acid sequence Tp of the target CA protein T.
The objective is being to locate a set of CA proteins, S = {S1, S2?} of similar sequence
? Select from S the primary structure Bp of a base CA protein, with a significant match to the target CA protein. A AIS-PSMACA [16],[18] search produces a measure of similarity between each CA protein in S and the target CA protein T. Therefore, Bp can be chosen as the CA protein with the highest such value
? Obtain the base CA protein"s structure, Bs, from the PDB
? Using Bp, create an input sequences Ib (corresponding to the base CA protein) by replacing each amino acid in the primary structure with its hydrophobia city value. The output sequences Ob is created by replacing the structural elements in Bs with the values, 200, 600, 800 for helix C, strand and coil respectively
? Solve the system identification problem, by performing CA de convolution with the output sequences Ob and the input sequence Ib to obtain the CA response, or the sought after running the algorithm.
?
7. Experimental Results
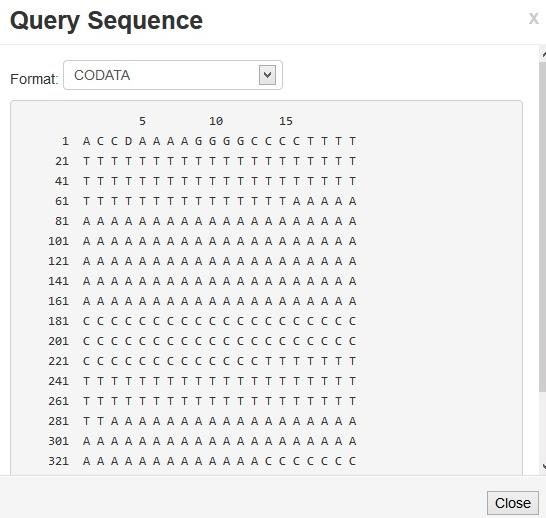
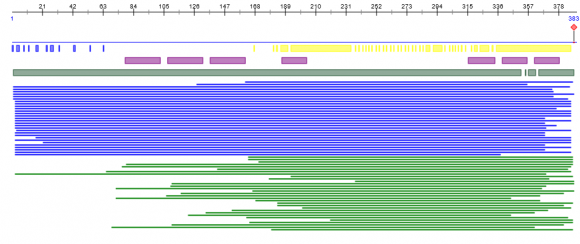
In the experiments conducted, the base proteins are assigned the values 300,700,900 for helix C, strand and coil respectively. We have found an structure numbering scheme that is build on Boolean characters of CA which predicts the coils, stands and helices separately. The MACA based prediction procedure as described in the previous section is then executed, and each occurrence of each sequences in the resulting output, is predicted. The query sequence analyzer was designed and identification of the green terminals of the protein is simulated in the figure 4. The analysis of the sequence and the place of joining of the proteins are also pointed out in the figure 5. Experimental results Figure 7, 8 which include the similarity and accuracy graph with each of the components are separately plotted.
8. Conclusion
Existing structure-prediction methods can predict the structure of protein with 75% accuracy. To provide a more thorough analysis of the viability of our proposed technique more experiments will be conducted .Our results indicate that such a level of accuracy is attainable, and can be potentially surpassed with our method. AIS-AIS-PSMACA provides the best overall accuracy that ranges between 80% and 89.8% depending on the dataset.
9. Global Journal of Computer Science and Technology
Volume XIII Issue IV Version I

| Target | |||||||
| : 1PFC | Prediction Accuracy | Target: 1PP2 | Prediction Accuracy | Target: 1QL8 | Prediction Accuracy | Year | |
| Exp 1 65% | Exp 5 | 85% | Exp 9 | 85% | |||
| Exp 2 65% | Exp 6 | 90% | Exp 10 | 90% | 15 | ||
| Exp 3 69% Exp 4 71% Prediction Method Prediction Accuracy for 1PFC Prediction Accuracy for Exp 7 83% Exp 11 Exp 8 87% Exp 12 1PP2 DSP 92% 70% PHD 70% 68% 68% 77% SAM-T99 | 82% 91% Prediction Accuracy 1QL8 96% 84% 87% | for | Volume XIII Issue IV Version I ( D D D D D D D D ) | ||||
| SS Pro AIS-PSMACA AIS-AIS-PSMACA | 70% 90% 92% | 73% 85% 83% | 81% 97% 96% | Global Journal of Computer Science and Technology | |||